暂无数据
使用基于片段的量子化学计算蛋白-配体相互作用能量的收敛协议。
原标题:Convergent Protocols for Computing Protein-Ligand Interaction Energies Using Fragment-Based Quantum Chemistry.
5 分
关键词
摘要
基于片段的量子化学方法提供了一种绕过电子结构计算陡峭非线性扩展的方法,从而可以使用高级方法研究大型分子系统。在这里,我们使用片段化来计算具有数千个原子的系统中的蛋白质-配体相互作用能,使用一个新的软件平台来管理基于片段的计算,该平台实现了筛选的多体展开。使用最小基组半经验方法(HF-3c)的收敛性测试表明,使用单残基片段和简单氢帽的双体计算足以再现使用传统超分子电子结构计算获得的相互作用能,误差在1 kcal/mol以内,而计算成本仅约为1%。我们还证明了HF-3c结果能够反映使用密度泛函理论在扩展四重-ζ质量基组中获得的趋势。战略性地部署片段化有助于在高质量电子结构方法和基组的支持下使用收敛的生物分子模型系统,将从头算量子化学引入到以前难以想象的大型系统中。这对于生成机器学习应用的高质量训练数据将是有用的。
AI理解论文
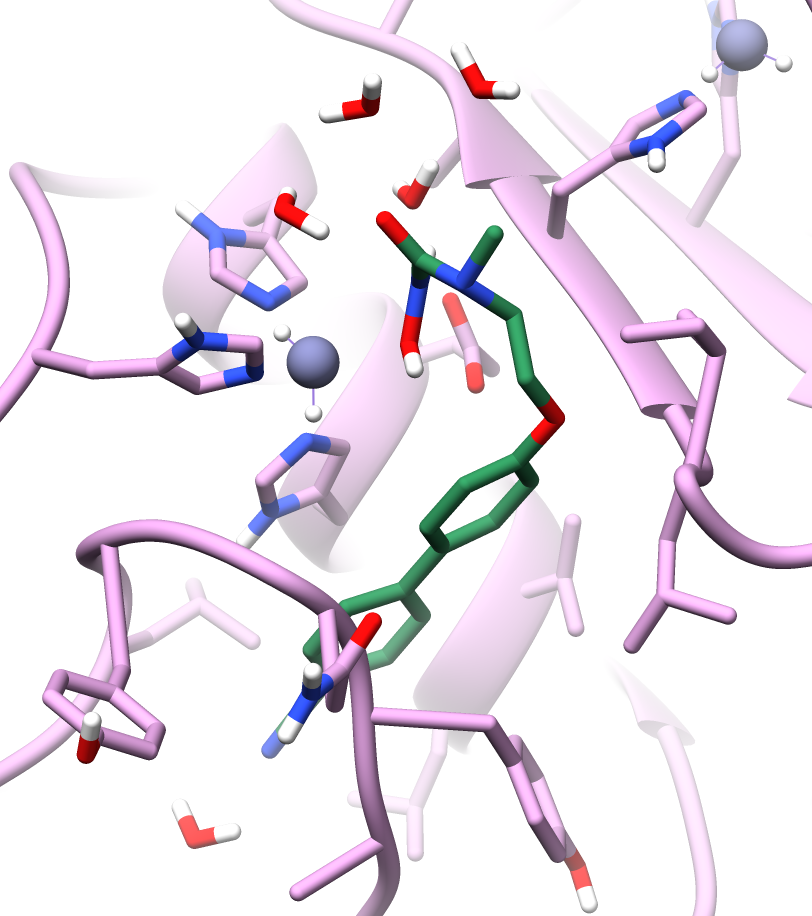
这篇论文主要探讨了一种新的碎片化方法,用于计算蛋白质-配体相互作用能量(E_int),并在电子结构理论的不同层次上进行研究。研究的重点是通过考虑多体相互作用和距离截断来提高计算的准确性和效率。以下是论文的主要内容和贡献:
研究背景与目标
论文的研究背景是蛋白质-配体相互作用在药物设计和生物化学中的重要性。传统的超分子计算方法虽然准确,但计算成本高昂,尤其是在处理大规模系统时。因此,研究的目标是开发一种系统可改进的碎片化协议,能够在高质量基组下进行计算,并在合理的计算成本下实现高精度。
方法与技术
-
碎片化方法:论文提出了一种基于碎片化的多体展开(MBE)方法,能够处理多达四体的相互作用。这种方法通过将大分子系统分解为较小的碎片来简化计算,并通过多体展开来考虑碎片之间的相互作用。
-
软件框架:研究中使用了一个新的软件框架Fragme\t,用于实现和测试所提出的碎片化方法。该框架支持不同的电子结构理论和基组,能够灵活地应用于各种蛋白质-配体系统。
-
计算精度:研究表明,对于小于20个原子的配体,所提出的方法能够在1,000+原子的酶模型中实现小于0.01 kcal/mol/碎片的碎片化误差。这一精度比传统的片段化方法提高了一个数量级。
实验与结果
-
小配体系统:在T4溶菌酶蛋白质系统中,研究验证了所提出方法的高精度,结果与传统的超分子计算方法一致。
-
大配体系统:对于更大的配体(如LIDS数据集),研究在某些情况下也能获得收敛的结果。然而,对于含有Zn2+的金属酶,由于金属离子附近的复杂相互作用,难以在合理的计算成本下实现高精度。
-
计算成本:与传统的超分子计算相比,所提出的方法在计算时间上具有显著优势,尤其是在使用DFT(密度泛函理论)层次的计算时。
讨论与未来工作
-
准确性与应用:论文讨论了在机器学习(ML)数据集生成中的应用潜力,认为即使在某些情况下未达到亚kcal/mol的精度,所提出的方法仍然足够用于生成可靠的ab initio数据集。
-
未来改进:未来的工作将考虑使用反作用点校正(counterpoise corrections)来进一步提高精度,并实施能量筛选以识别重要的三体相互作用。
-
挑战与局限:研究指出,尽管方法在小配体系统中表现良好,但在处理更复杂的系统(如含金属离子的酶)时仍面临挑战。
结论
论文的主要贡献在于开发了一种系统可改进的碎片化方法,能够在高质量基组下实现高精度的蛋白质-配体相互作用能量计算。这一方法在计算效率和精度上均优于传统方法,具有广泛的应用潜力,尤其是在药物设计和生物化学计算中。
通过这篇论文,研究者为大规模计算生物化学应用中的电子结构理论计算提供了一种新的工具,展示了其在处理复杂生物分子系统中的潜力和优势。