暂无数据
蛋白质语言可视化工具:语言模型时代的序列相似性网络
原标题:The Protein Language Visualizer: Sequence Similarity Networks for the Era of Language Models
5 分
关键词
摘要
高通量测序技术的出现和生物“大数据”的可用性加速了新蛋白质序列的发现,使得跟上其功能注释的步伐变得具有挑战性。为了解决这一注释挑战,诸如序列相似性网络(SSNs)等技术被用于可视化地分组蛋白质以加快识别速度。在本文中,我们介绍了一种替代的可视化分析工具。我们简单且模块化的蛋白质语言可视化器(PLVis)管道结合了预训练的PLM嵌入、降维算法和聚类方法,以便根据邻居快速评估蛋白质。生成的交互式可视化可以通过多层生物和结构注释进行丰富。我们对PLVis与SSNs进行了正面对比,发现了一致的聚类模式,PLVis提供了额外的优势,即揭示了在SSN中看似孤立节点的蛋白质之间的关系。接下来,我们定量验证了这样一种直观概念:在PLVis中分离良好的簇比投影中大的、中心的“模糊”区域更忠实地捕捉序列相似性信息,在这些区域中,相对距离的解释应谨慎。此外,我们展示了一些案例研究,证明了该管道在探索多生物体全蛋白质组比较中的价值。最后,我们通过共享PLVis Colab Notebook,使这一管道对更广泛的研究社区可访问。PLVis为研究人员提供了一个低门槛的平台,通过语言模型的视角来探索和比较蛋白质组。
AI理解论文
这篇论文介绍了一种新的蛋白质序列分析工具,称为PLVis,它利用**蛋白质语言模型(PLM)的嵌入技术来增强蛋白质序列的功能注释。随着高通量测序技术的发展,蛋白质序列的发现速度大大加快,但功能注释的速度却未能跟上。传统的序列相似性网络(SSN)**通过可视化蛋白质之间的关系来帮助解决这一问题,但存在一些局限性。PLVis通过使用PLM嵌入和降维算法来克服这些局限性,提供了一种更为直观和交互的分析方法。
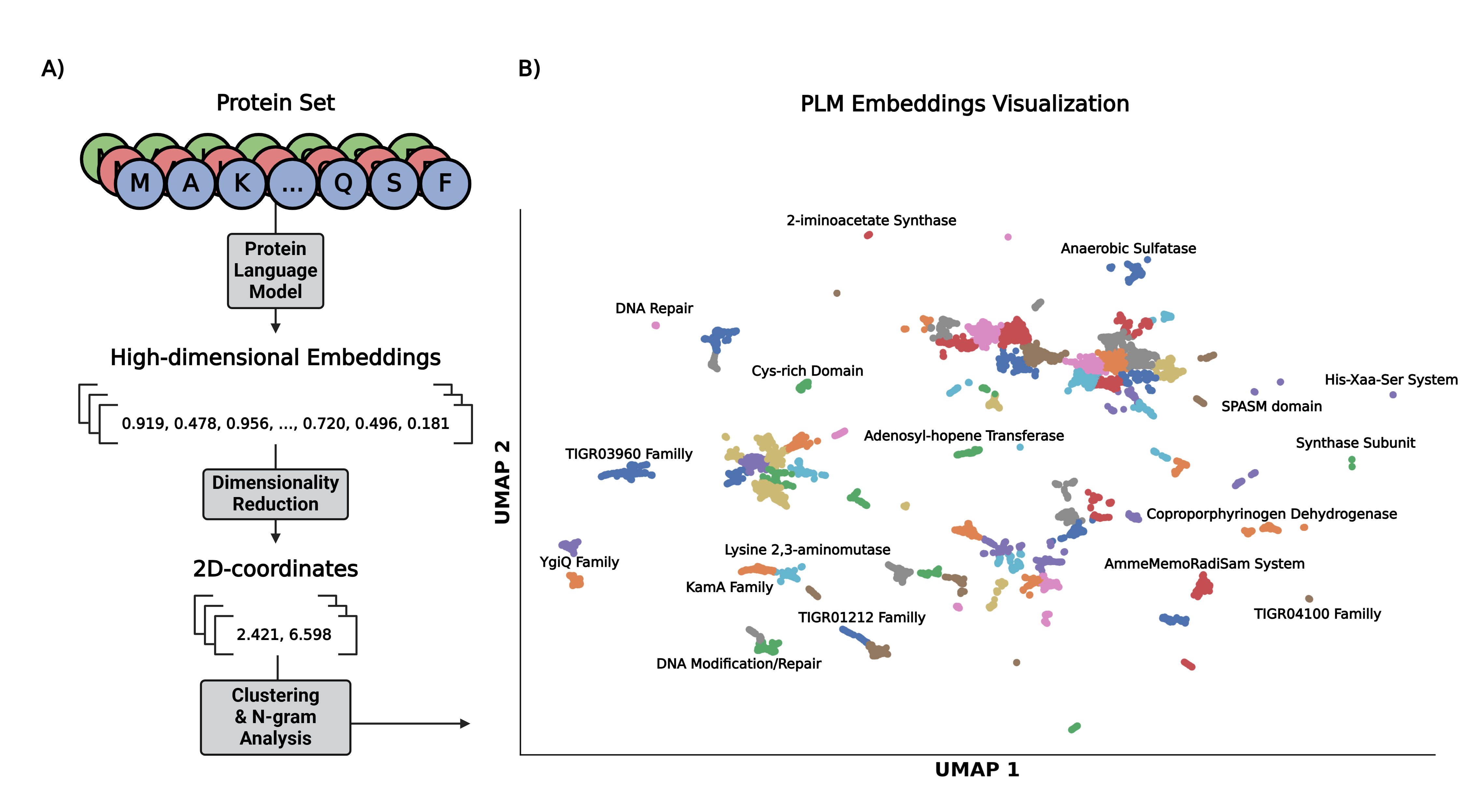
PLVis管道的工作流程包括以下几个步骤:首先,将一组蛋白质序列输入到PLM中以获得高维嵌入。然后,使用降维算法将这些嵌入减少到二维。最后,对数据进行聚类,并通过n-gram分析生成适当的聚类标题以完成可视化。该管道的模块化设计允许用户选择不同的模型进行嵌入生成、降维和聚类。
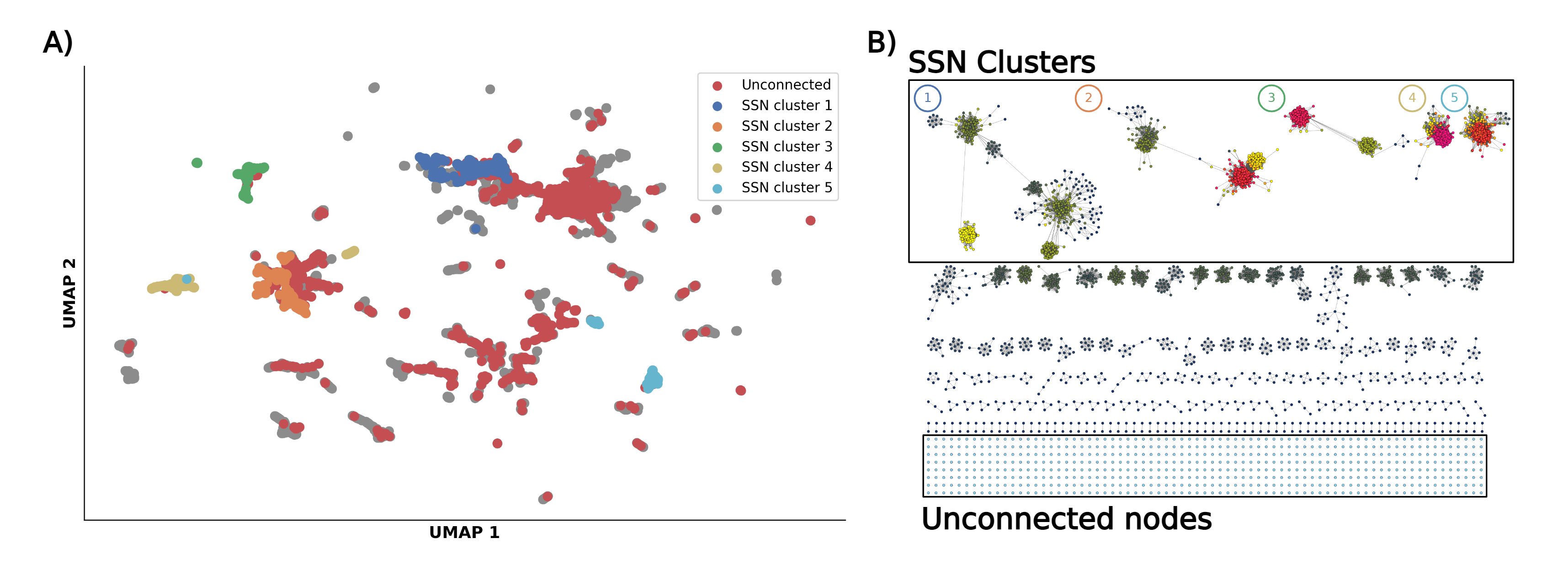
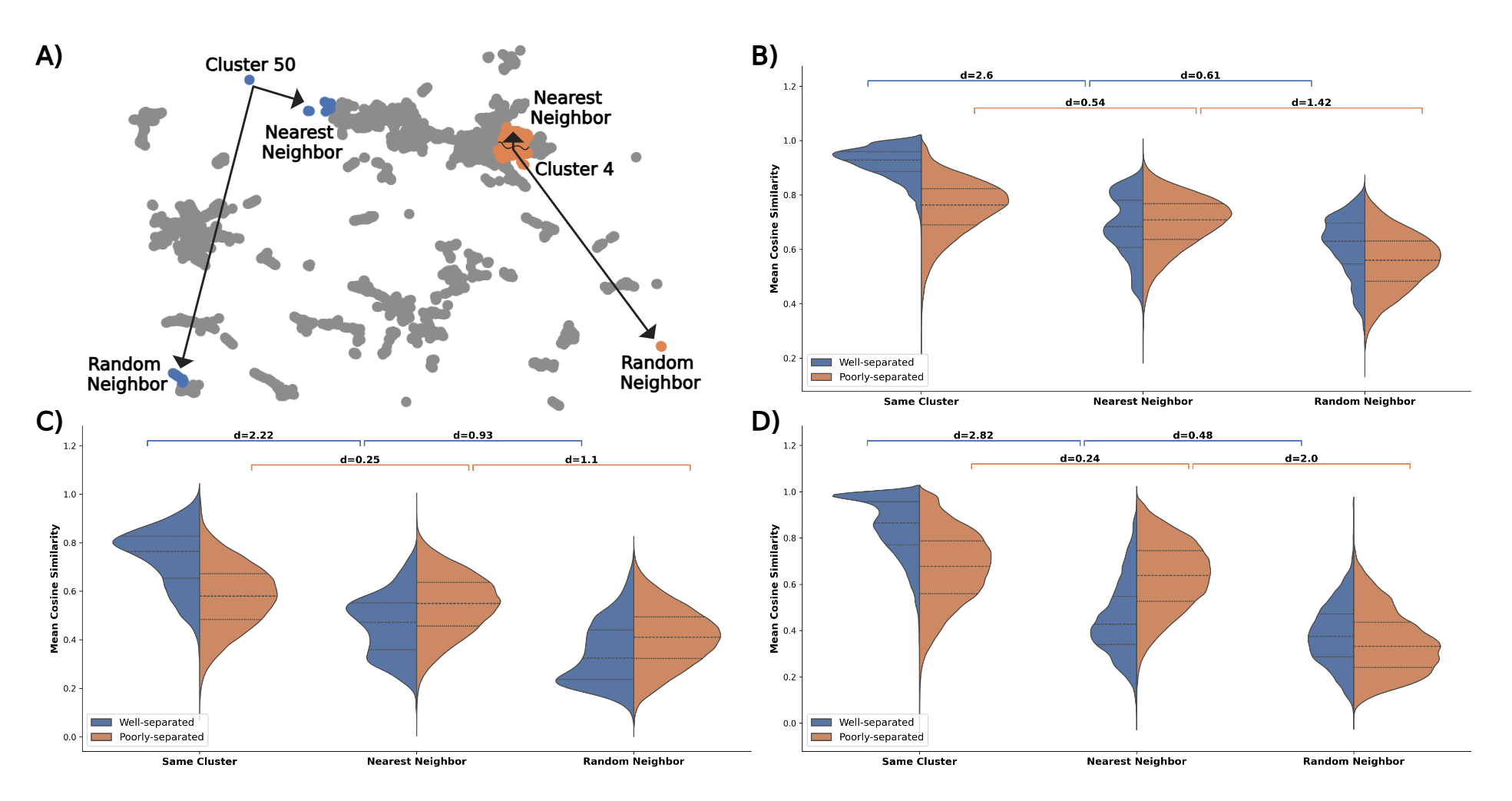
论文中对PLVis和SSN进行了比较分析。SSN依赖于用户设定的阈值来创建节点之间的边,这个阈值对应于序列比对得分。过高的阈值可能导致许多孤立的蛋白质,而过低的阈值可能导致过拟合。PLVis通过PLM嵌入和降维技术,能够在不依赖于二元阈值的情况下,识别蛋白质的聚类关系。研究中使用了10,000个随机选择的rSAM酶进行比较,结果显示PLVis能够更好地保留高维信息,并在二维空间中形成清晰的聚类。
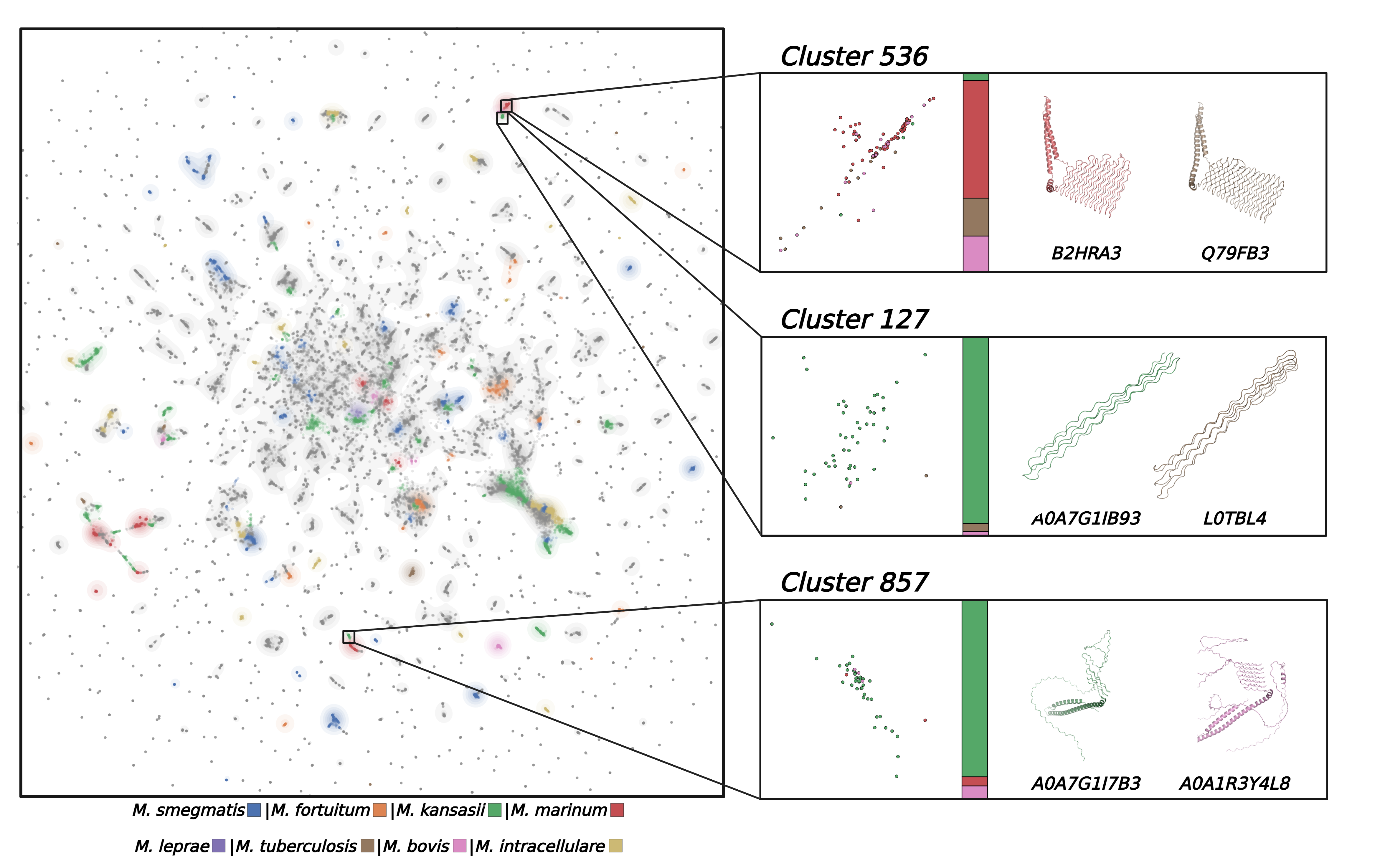
PLVis的优势在于其能够识别出在SSN中被孤立的蛋白质,并将其与具有相似功能的蛋白质进行关联。例如,研究发现,PLVis中的某些聚类能够将之前在SSN中未连接的蛋白质与共享相同InterPro“家族”条目的其他蛋白质关联起来。这表明PLVis在识别潜在功能相关的蛋白质群体方面具有重要价值。
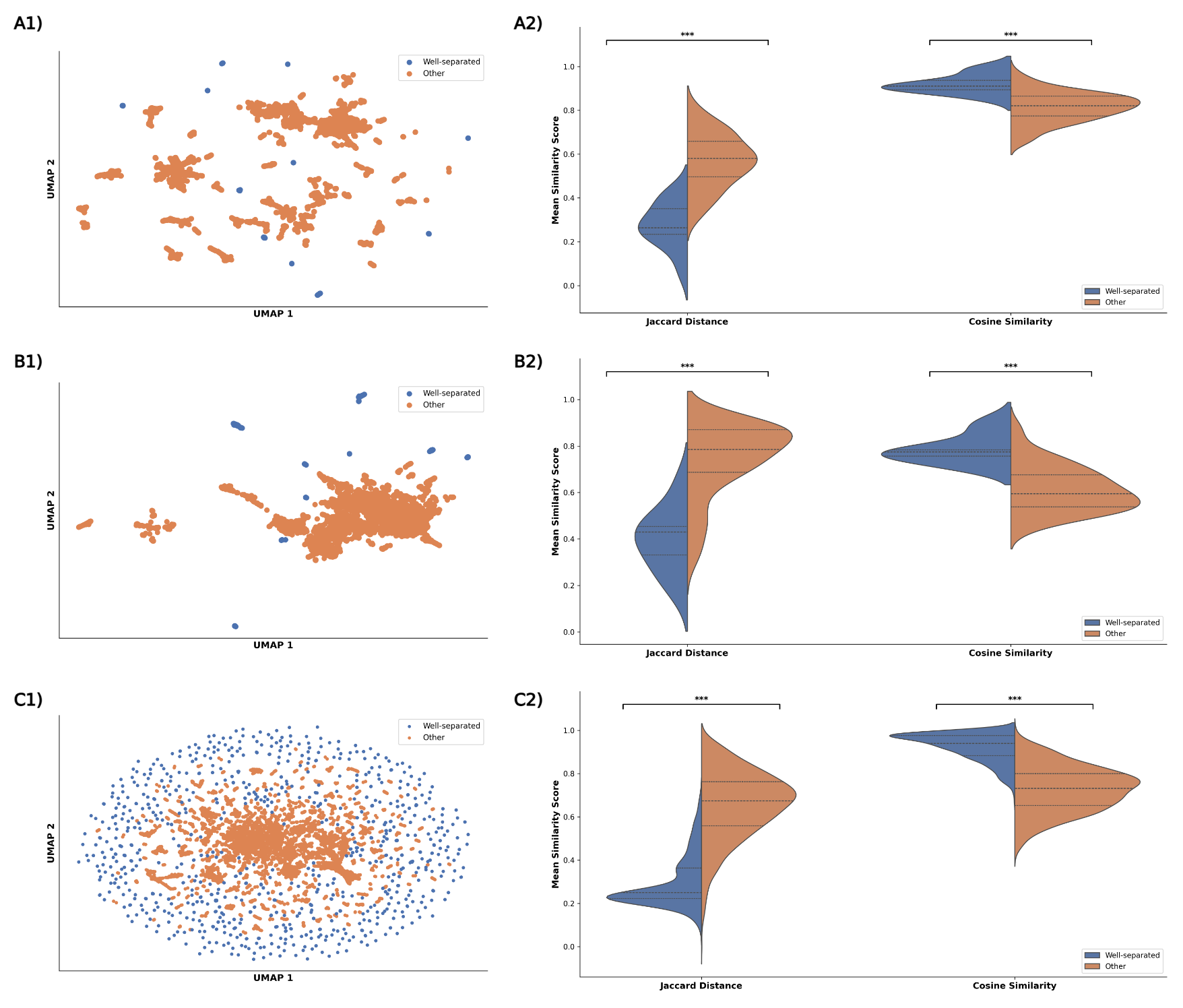
此外,论文还探讨了PLVis在保留高维嵌入信息方面的能力。通过计算Jaccard距离,研究评估了PLM嵌入的二维投影是否能够保持高维信息。结果表明,分离良好的聚类能够更好地反映高维空间中的邻近关系,而大型、中心化的聚类则往往表现出较差的邻近关系。
PLVis的应用不仅限于个体蛋白质分析和聚类组织,还在更广泛的比较研究中展示了显著的实用性。通过比较不同物种的完整蛋白质组,PLVis能够揭示重要的生物学见解,如物种特异性蛋白质家族的缺失或在分类学属内的保守模式。这种方法在分析特定生物学关系(如宿主-病原体相互作用)时尤为有价值。
为了促进PLVis的广泛应用,研究团队提供了一个公共的Google Colaboratory笔记本,供研究人员探索他们的蛋白质数据集。这一工具为复杂的蛋白质组数据集的可视化探索和分析提供了一个强大的框架,填补了高通量生物数据解释中的关键空白。
总之,PLVis通过结合PLM嵌入和降维技术,为蛋白质序列的功能注释提供了一种创新的解决方案。它不仅克服了传统SSN的局限性,还为蛋白质组学研究提供了新的视角和工具。研究团队鼓励更广泛的科学界在其研究工作流程中评估和实施PLVis,并积极寻求社区反馈和贡献,以扩展其分析能力和应用。通过迭代开发和协作改进,PLVis旨在成为现代生物信息学工具包的重要组成部分,促进对蛋白质组数据分析的深入理解。