暂无数据
SeqLengthPlot:一个易于使用的基于Python的工具,用于可视化和检索fasta文件中的序列长度,并具有可调的分割点。
原标题:SeqLengthPlot: An easy-to-use Python-based Tool for Visualizing and Retrieving Sequence Lengths from fasta files with a Tunable Splitting Point
5 分
关键词
摘要
动机 精确的序列长度分析在生物信息学中尤为重要,特别是在基因组学和蛋白质组学领域。现有的工具如SeqKit和Trinity工具包等提供基本的序列统计,但在提供全面的分析和绘图选项方面往往有所不足。例如,SeqKit是一个非常完整且快速的序列分析工具,能够提供有用的指标(如序列数量、平均长度、最小长度、最大长度),并可以返回给定长度范围内较短或较长的序列(单侧,不是同时两侧)。类似地,Trinity的实用珍珠脚本提供详细的重叠群长度分布(如N50、中位数和平均长度),但不包括序列总数,也不提供数据的图形表示。结果 鉴于关键的序列分析任务分布在不同的工具中,我们推出SeqLengthPlot:一个易于使用的基于Python的脚本,填补了生物信息学工具在序列长度分析方面的空白。SeqLengthPlot生成全面的统计摘要,从输入的FASTA(核苷酸和蛋白质)文件中根据可调的用户定义的序列长度过滤并自动检索序列到两个不同的文件中,并生成相应序列的图表或动态可视化。可用性和实现 详细的SeqLengthPlot流程在GitHub上提供,网址为https://github.com/danydguezperez/SeqLengthPlot,遵循GPL-3.0许可证发布。作为来源或用例编译的其他数据集可通过Mendeley Data存储库公开获取:DATASET_Ss_SE.1: http://dx.doi.org/10.17632/pmxwfjyyvy.1, DATASET_Ss_SE.2: http://dx.doi.org/10.17632/3rtbr7c9s8.1, DATASET_Ss_SE.3: http://dx.doi.org/10.17632/wn5kbk5ryy.1, DATASET_Ss_SE.4: http://dx.doi.org/10.17632/sh79mdcm2c.1 和 DATASET_Ss_SE.5: http://dx.doi.org/10.17632/zmvvff35dx.1。
AI理解论文
该文档介绍了一种名为SeqLengthPlot.py的Python脚本工具,旨在改进FASTA文件中序列长度的分析和可视化。以下是对该文档的详细总结:
背景与动机
在基因组学和蛋白质组学中,准确的序列长度分析至关重要。现有工具如SeqKit和Trinity工具包提供了一些基本的序列统计功能,但在综合分析和绘图选项方面存在不足。例如,SeqKit能够快速提供序列分析的基本指标(如序列数量、平均长度、最小和最大长度),但无法同时返回两侧的序列长度范围。Trinity的工具则提供了详细的contig长度分布(如N50、中位数和平均长度),但不包括序列总数,也不提供数据的图形表示。因此,SeqLengthPlot的开发旨在填补这些工具在序列长度分析中的空白。
工具描述
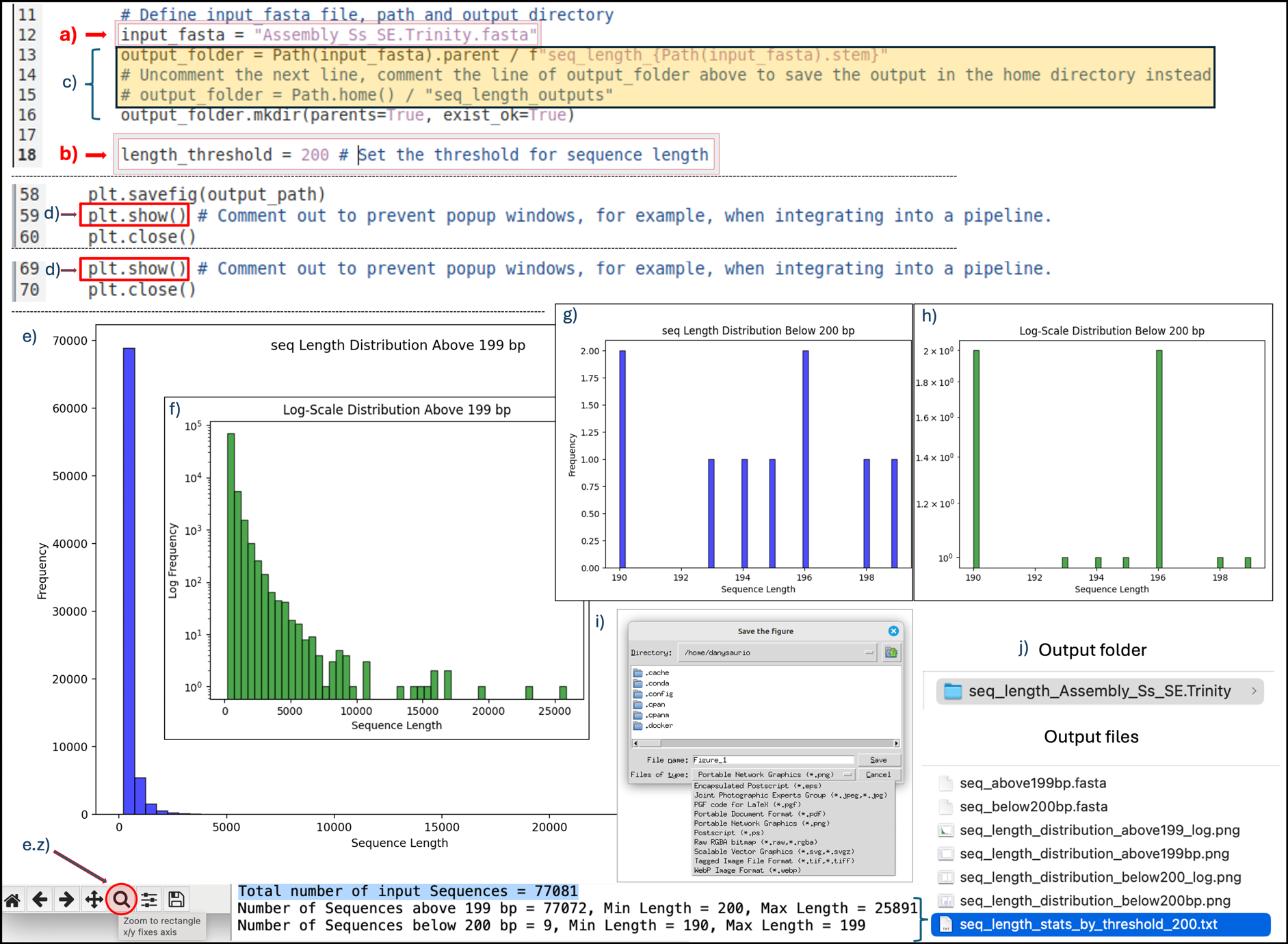
SeqLengthPlot.py是一个易于使用的Python脚本,专为增强序列长度分析而设计。该脚本从FASTA文件中处理序列数据,以对转录本长度进行分类和分析。它生成指定阈值以上和以下的转录本长度的直方图,并提供这些分布的统计摘要。
- 输入处理:脚本从指定的核苷酸或蛋白质FASTA文件中读取序列。
- 输出目录管理:默认情况下,输出文件保存在与输入FASTA文件相同的目录中。
- 长度阈值:序列根据脚本中设置的长度阈值(默认200个碱基对)进行分类。
- 图表生成:生成序列长度的直方图,并导出为线性和对数刻度的图表。
- 统计输出:计算并保存输入和结果FASTA文件的序列长度统计信息。
应用
SeqLengthPlot特别适用于以下方面的评估:
- 转录组序列长度截止准确性:评估使用RNAseq组装器(如Trinity)时所用标准截止长度的准确性。
- ORF和肽段长度的探索:分析由Transdecoder或其他工具生成的ORF的出现和分布。
兼容性、安装和依赖
该工具兼容Unix和Windows操作系统。运行SeqLengthPlot.py需要安装Python及其库:matplotlib、Biopython和Pathlib。
推荐实施步骤
- 下载脚本:从GitHub下载SeqLengthPlot.py脚本,并将其放置在包含输入FASTA文件的文件夹中。
- 设置参数:
- 定义路径:修改脚本中的输入FASTA文件路径。
- 定义序列长度:设置所需的长度截止值。
- 可选参数:更改输出路径或禁用交互式图表显示。
- 运行脚本:在终端或命令提示符中导航到包含SeqLengthPlot.py的文件夹,然后执行脚本。
输出文件
- 直方图图表:生成四个PNG文件,显示序列长度的直方图。
- FASTA文件:根据长度阈值将序列分为两部分,并分别输出为FASTA文件。
- 统计摘要:生成一个文本文件,包含输入FASTA的序列长度详细统计信息。
结论
SeqLengthPlot.py将多种功能整合到一个高效的平台中,使其成为增强FASTA文件序列长度分布评估的有用工具,包括绘图、过滤和自动序列检索。
贡献与可用性
该工具通过提供综合统计摘要、过滤和自动序列检索,填补了现有生物信息学工具在序列长度分析中的空白。SeqLengthPlot的详细管道可在GitHub上获取,并在Mendeley Data存储库中提供了额外的数据集作为使用案例。
通过以上总结,读者可以全面理解SeqLengthPlot.py的内容、方法和贡献。该工具在生物信息学领域提供了一种有效的解决方案,用于序列长度的分析和可视化。